Cardiovascular Disease

Clinical Summary from Dr David Samra
Dr David Samra is a Fellowship-trained Sport and Exercise Medicine Physician with clinical experience focused on prevention of cardiovascular disease in both athletes and the general population.
Atherosclerotic cardiovascular disease (ASCVD) remains the dominant limiter of lifespan and healthspan in Western populations. Across epidemiology, guidelines, and mechanistic biology, the data converges on the same point: cardiovascular events are late expressions of a disease process that begins decades earlier and accumulates silently. The clinical implication is significant. If we keep using risk tools designed to predict 10-year events, we will continue to miss disease that has been building for 30 years. This article works through the biology, the evidence, and the clinical framework that has shaped my approach at Progressive Longevity Clinic.
Last reviewed: May 2026
Author: Dr David Samra, MBBS (Hons), MD, FACSEP – Sport and Exercise Medicine Physician
Interested in evidence-based cardiovascular risk assessment that goes beyond the standard lipid panel?
At Progressive Longevity Clinic, we work through ApoB, Lp(a), metabolic accelerators, and plaque imaging where appropriate — with proportionate, individualised intervention.
Visit our website to learn more about our Sport & Exercise Specialist-Led Longevity Medicine approach.
How my thinking changed...
I did not train intending to practise longevity medicine. My background is in Sport and Exercise Medicine — managing weight, insulin resistance, metabolic disease, and musculoskeletal pathology within standard guideline frameworks. Like most clinicians, I treated lipids in line with consensus targets and 10-year risk calculators.
My interest deepened through early exposure to low-carbohydrate approaches and diabetes management, and then through real-world clinical observation: improvements in pain, osteoarthritis progression, and metabolic markers that were not easily explained by weight loss alone. Patients were doing better than I expected, and the conventional explanations did not fully account for what I was seeing.
A major influence was Peter Attia — not because he claims to have answers, but because he consistently asks better questions of people who understand biology deeply. His interviews led me back into lipidology, and particularly into Tom Dayspring’s work, which clarified how much of what we call “normal” is simply population-based normality, not biological safety. The framework that follows is the endpoint of that curiosity — reading the primary literature, listening to people who think rigorously, and applying it carefully in clinical practice.
ASCVD Dominates Lifespan and Healthspan
If we step back and look at mortality, morbidity, and quality-adjusted life years, ASCVD still dominates. Across epidemiology, guidelines, and mechanistic biology, it remains the principal limiter of both lifespan and healthspan in Western societies. Cancer comes second. Everything else trails further behind.
The conceptual shift for me was reframing ASCVD as an exposure disease, not an event disease. Events are late. The biology starts early. That framing appears consistently in Mendelian randomisation data, lipidology literature, and long-term cohort studies. Once you accept that framing, it becomes difficult to justify late, threshold-based intervention as an optimal strategy.
Mendelian randomisation studies are particularly informative here. Genetic variants that lower LDL-C from birth confer cardiovascular risk reductions that are roughly three-fold greater per mmol/L than the same magnitude of LDL-C reduction achieved by statins started in midlife [1]. The biology rewards earlier exposure to lower particle burden disproportionately. That alone should reshape how we think about prevention.
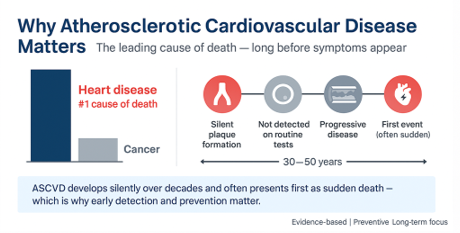
A Long Disease Measured With Short Tools
ASCVD evolves silently over 30 to 50 years, yet we predominantly assess it using tools designed to predict 10-year events. That mismatch matters.
I do not think clinicians are doing anything wrong. The tools we are given are limited by design. Ten-year risk calculators are useful for deciding who qualifies for statins today within current guidelines. They are not designed to identify cumulative disease burden in a 40-year-old with normal LDL-C, a strong family history, and slowly progressing insulin resistance. That is a different question entirely.
Once you accept that ASCVD burden scales with atherogenic particle exposure over time, reassuring a 40- or 50-year-old on the basis of low short-term risk becomes biologically uncomfortable. The logical question then becomes: what should we be measuring, and how early should we intervene?
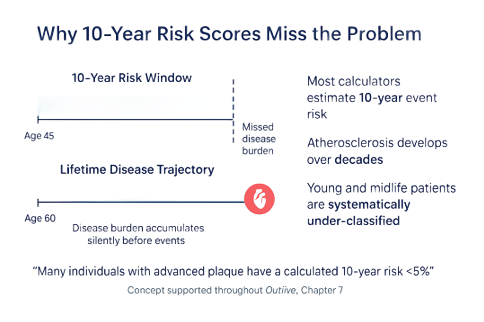
Risk Is Cumulative, Not Binary
ASCVD risk is not binary, and it is not sudden. It is cumulative exposure. This is not an argument for treating every 30-year-old pharmacologically — it is an argument for understanding risk in a time-dependent way and intervening proportionately
Peter Attia’s framing is helpful: risk is not cholesterol today — it is cholesterol multiplied by years of exposure. The biological substrate accumulates. The question for any individual patient becomes how much of that accumulating exposure is biologically warranted, and where the curve can be safely shifted.

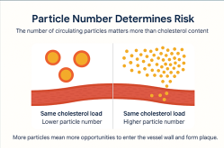
The data on this is robust. Combined Mendelian randomisation analyses show that lifetime exposure to lower LDL-C and lower systolic blood pressure produces dramatic reductions in major vascular events — effects far greater than what is achievable with late-onset pharmacological intervention [2]. The biology is doing arithmetic over decades; our risk models are doing arithmetic over a single decade.
Cholesterol Is Not the Causal Agent
For decades, we have spoken about cholesterol as though it is the causal agent in atherosclerosis. The simplification has been useful for population health messaging, but it obscures the underlying biology.
When you engage with the biology, cholesterol itself is not what initiates plaque formation. The causal agents are the apoB-containing lipoprotein particles that transport cholesterol through the bloodstream — LDL, VLDL, IDL, and Lp(a). Every atherogenic particle has the opportunity to interact with the artery wall. Risk is therefore a function of particle number and duration of exposure, not cholesterol content alone.

This explains why patients with identical LDL-C can have very different disease trajectories. One patient may have fewer particles each carrying more cholesterol; another may have many smaller particles each carrying less cholesterol. The cholesterol number is the same. The risk profile is not.
LDL-C is a useful approximation. ApoB is the actual question. When the two disagree, ApoB is the signal to trust.
ASCVD Is a Disease of the Artery Wall
Ultimately, ASCVD is a disease of the artery wall — specifically of the endothelium. The endothelium is one cell thick, but it is not a passive lining. It is metabolically active, regulating tone, inflammation, coagulation, and permeability.
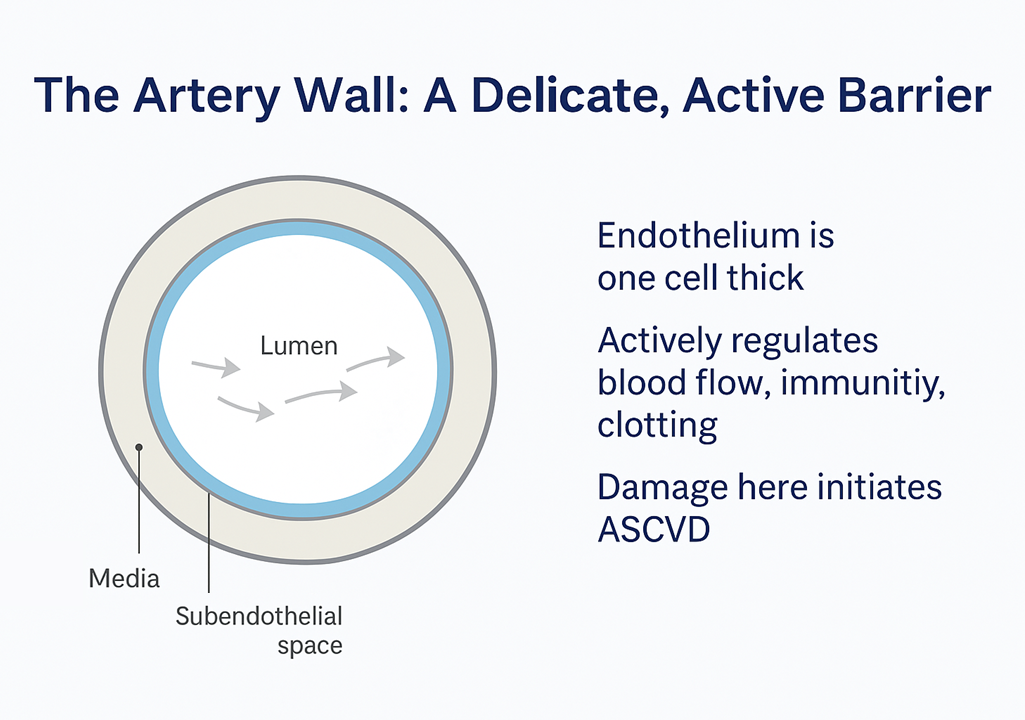
ApoB tells us how many particles are knocking on the door. Endothelial health determines how often the door opens. This is where lipidology and metabolic health converge into a single biological story — and why insulin resistance, visceral adiposity, hypertension, and chronic inflammation all amplify the damage that any given particle burden can cause.
Cholesterol Is Cargo, Not the Vehicle: ApoB as Particle Count
Cholesterol does not circulate freely in the bloodstream — it is hydrophobic and must be transported within lipoprotein particles. Those particles differ structurally and biologically. HDL and LDL may both carry cholesterol, but they behave very differently in the vasculature.
When we measure cholesterol, we measure cargo. Risk is determined by the vehicle. Separating cholesterol from its carriers resolves much of the confusion in lipid medicine.
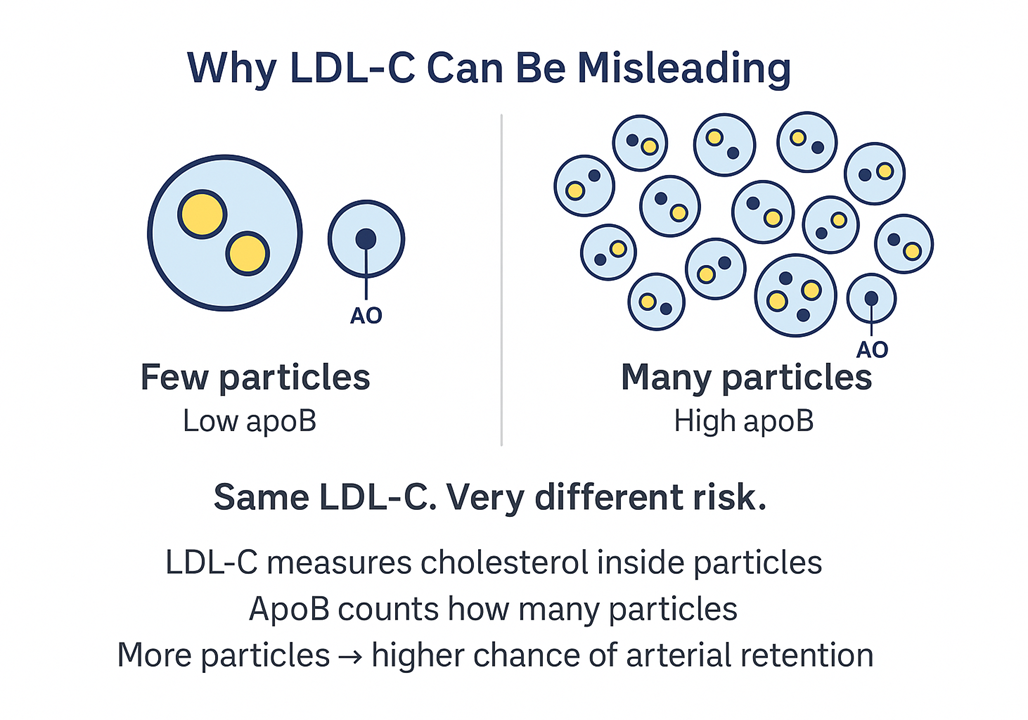
Every atherogenic particle — LDL, VLDL, IDL, and Lp(a) — carries exactly one apoB molecule. There are no exceptions. ApoB therefore directly reflects particle number, independent of cholesterol content per particle. This is what makes apoB such a clean clinical marker: one apoB equals one atherogenic particle [3].

The Marston analysis from the TIMI Study Group — published in JAMA Cardiology in 2022 — is particularly important here. Across nearly 400,000 individuals in primary prevention and a separate secondary prevention cohort, apoB was the only lipid parameter significantly associated with myocardial infarction risk after adjustment. Neither LDL-C nor non-HDL cholesterol added information beyond what apoB provided. For a given apoB level, particle type and cholesterol content did not change risk [4]. The implication is that apoB tells you essentially everything that matters about atherogenic particle-related risk — once you know the particle count, the rest is detail.
ASCVD risk tracks with cumulative apoB exposure multiplied by time. That single sentence reframes most of clinical lipid medicine.
Discordance Explains the “Normal Cholesterol” Heart Attack
When LDL-C and apoB are concordant — both high, both low — either marker tracks risk reasonably well. When they are discordant, apoB outperforms LDL-C consistently across discordance studies.
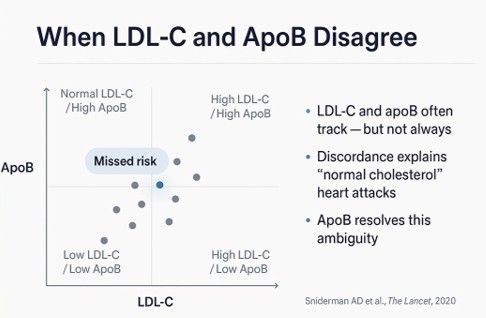
Discordance is common, particularly in insulin resistance, hypertriglyceridaemia, and certain dietary states. The classic pattern is normal LDL-C with elevated apoB — many small dense LDL particles each carrying less cholesterol. The cholesterol number looks reassuring; the particle count is anything but. This explains the clinical scenario we have all encountered: the so-called “normal cholesterol” myocardial infarction.
When the two disagree, apoB is the signal to trust. This is not an opinion — it is the consistent finding across discordance analyses, Mendelian randomisation studies, and the European Society of Cardiology / European Atherosclerosis Society 2019 guidelines, which formally elevated apoB to the recommended primary measure of atherogenic lipoprotein risk.
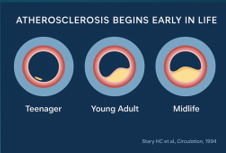
Atherosclerosis Begins Early in Life
Pathology data — from the foundational work of Stary and colleagues onward — demonstrates that atherosclerosis begins early, often in adolescence. Autopsy studies in young trauma victims have shown fatty streaks and early lesions in the second and third decades of life, well before any clinical disease is recognisable.
This is not an argument for medicating children. It is an argument for understanding trajectory, biology, and exposure long before clinical disease manifests. That concept underpins the entire longevity-oriented approach to ASCVD: we are working with disease that is already biologically present in many of our middle-aged patients, even when they look and feel well.
Retention Is the Initiating Event
ApoB-containing particles cross the endothelium routinely — this alone is normal physiology. The endothelium is not impermeable; particles transit it constantly as part of normal lipid metabolism. Pathology begins when particles are retained in the subendothelial space.
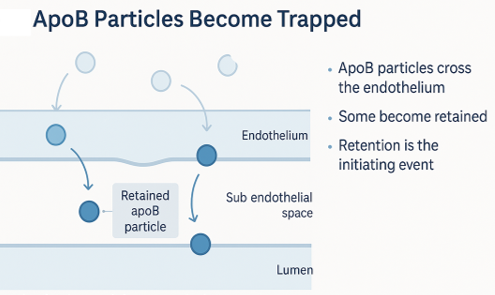
Retention initiates oxidation, immune activation, foam cell formation, and ultimately plaque development. The seminal mechanistic work — articulated most clearly in the response-to-retention hypothesis by Williams and Tabas — reframed atherosclerosis as a disease driven by particle trapping rather than by passive cholesterol deposition. This is now consensus mechanism.
This is paragraph text. Click it or hit the Manage Text button to change the font, color, size, format, and more. To set up site-wide paragraph and title styles, go to Site Theme.
Retention — not rupture or thrombosis — is the initiating event in atherogenesis. Everything downstream is a consequence.
From Retention to Plaque Formation
Once apoB particles are retained in the subendothelial space, the cascade is biologically predictable. The particles are oxidised and otherwise modified, becoming biologically toxic. Monocytes are recruited, differentiate into macrophages, and engulf the modified particles — becoming the foam cells that define the early atherosclerotic lesion.
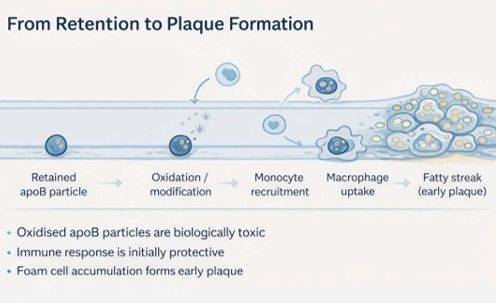
With chronic exposure, macrophage clearance becomes overwhelmed. Foam cells accumulate. Fatty streaks develop. The immune response, which was initially protective, becomes part of the pathological process. This is silent, decades-long disease that precedes any luminal narrowing.
Peter Libby’s 2021 Nature review captures the modern synthesis well: atherosclerosis is fundamentally an inflammatory disease driven by lipid retention, in which the immune response is both cause and consequence of the lesion [5]. By the time stenosis or plaque rupture occurs, disease has been biologically established for a long time. This is why early intervention matters — because by the time events occur, we are intervening at the end of a long biological story, not the beginning.
Why 10-Year Risk Calculators Underestimate Disease
Ten-year risk tools perform adequately in older, high-risk populations — the patients for whom they were primarily developed and validated. They fail in younger and midlife patients because ASCVD is not a 10-year disease.
The calculator is not wrong. It is answering a short-term event question, not a long-term disease question. A 45-year-old with normal LDL-C, no diabetes, and a normal blood pressure may have a calculated 10-year risk under 5% — and may also have advanced subclinical atherosclerosis on coronary imaging. The two findings are not contradictory; they reflect that the calculator is asking a different question than the imaging is answering.
Once you understand this, you stop using 10-year risk as reassurance. It becomes one input in a broader framework that includes apoB, Lp(a), metabolic health, family history, and — when appropriate — imaging. Reassurance based on a 10-year calculator alone is biologically misleading.
ApoB Simplifies Risk Assessment
ApoB answers a simple question: how many atherogenic particles are circulating? It captures both LDL-related and VLDL-related risk in a single measurement, performs better than LDL-C in metabolic disease, and consistently outperforms LDL-C in risk discrimination across the populations that have been studied.
The Sniderman 2019 narrative review in JAMA Cardiology synthesises the evidence well: across prospective epidemiological studies, randomised trials, and Mendelian randomisation analyses, apoB more accurately reflects cardiovascular risk than LDL-C or non-HDL cholesterol [6]. The real debate now is not about whether apoB is useful — that question is settled. The debate is about timing, thresholds, and which patients warrant intervention based on apoB alone.
Pragmatically, this is not difficult to operationalise. ApoB is widely available, inexpensive, requires no special preparation beyond a standard fasting lipid panel, and is reproducible across labs. There is no good clinical reason for it not to be a routine part of cardiovascular risk assessment in 2025.
Lp(a): The Hidden Genetic Risk Factor
Lipoprotein(a), or Lp(a), is genetically determined, pro-atherogenic, and pro-thrombotic. Structurally, it is an LDL-like particle with an additional apolipoprotein(a) molecule covalently bound to the apoB-100 backbone. The particle behaves like LDL in terms of arterial retention but with additional thrombogenic and inflammatory properties.
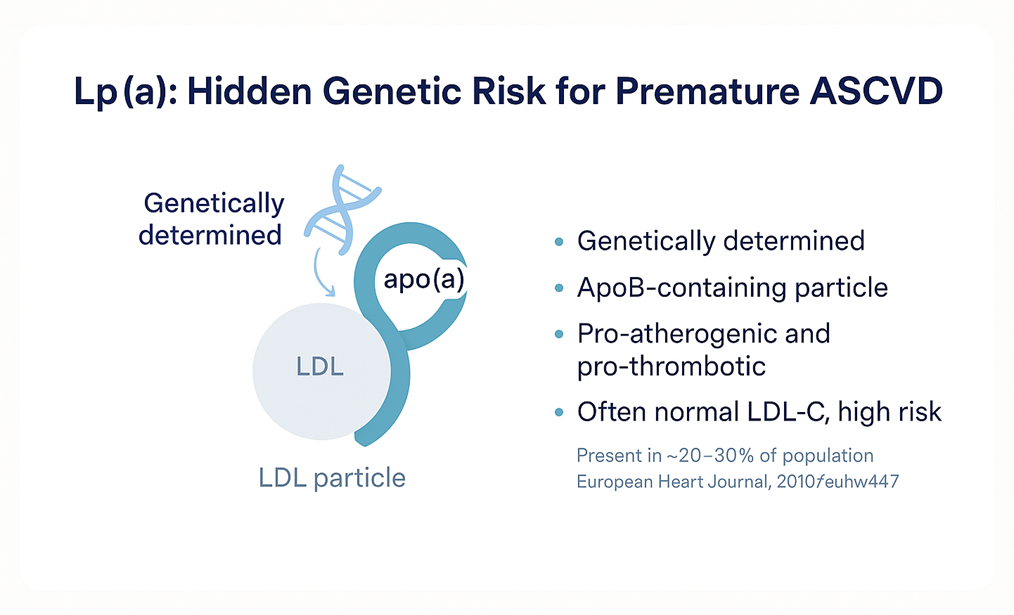
Approximately 20–30% of the population has elevated Lp(a). LDL-C may be normal yet lifetime cardiovascular risk substantially elevated. Levels are around 90% genetically determined and remain stable across the lifespan, which makes Lp(a) a once-in-a-lifetime test — measure it once, and you have the answer for that person’s lifetime [7].
When elevated, current management focuses on reducing total cumulative apoB exposure — because while we cannot yet meaningfully lower Lp(a) itself with available therapies, we can reduce the broader atherogenic particle burden against which Lp(a) is acting. New Lp(a)-specific therapies (antisense oligonucleotides and small interfering RNAs) are in late-stage trials and may change this landscape within the next few years. For now, identification matters because it shifts the threshold for proportionate intervention.
Advanced Lipid Testing as Adjuncts
Beyond apoB and Lp(a), more granular lipid testing exists — LDL subfraction analysis, sterol panels (lathosterol, desmosterol, sitosterol). These do not replace apoB. They are adjunctive tools used selectively to understand mechanism, discordance, or therapeutic response.
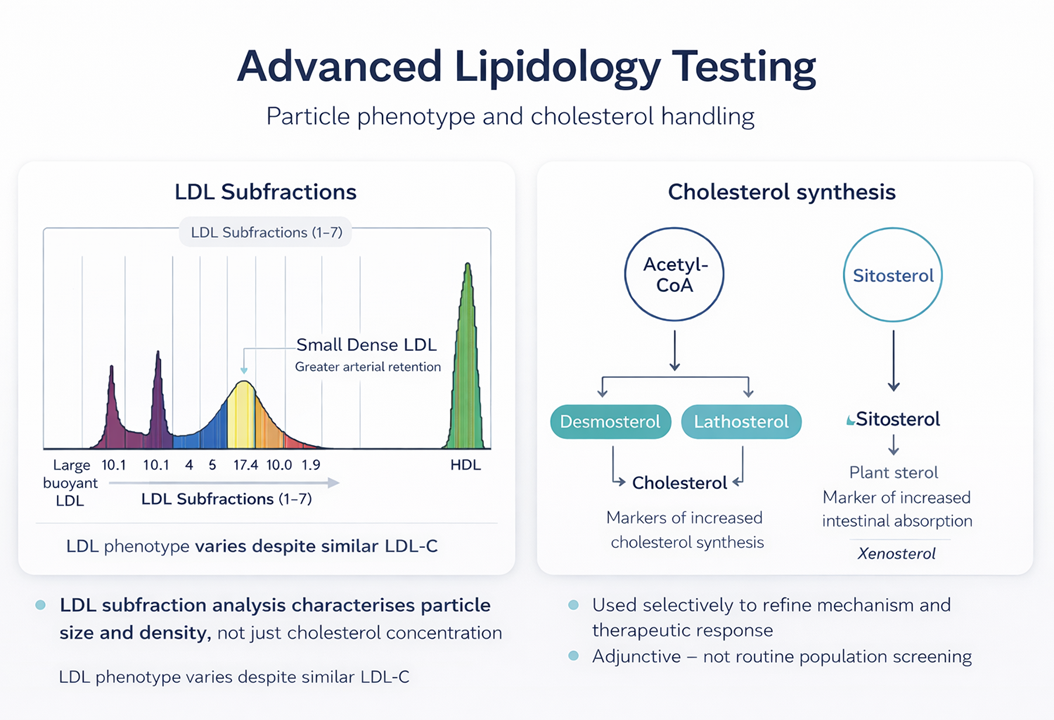
LDL subfraction analysis can tell us whether a patient’s particles are predominantly large and buoyant or small and dense — with small dense LDL associated with greater arterial retention. Sterol testing can identify whether dyslipidaemia is being driven primarily by hepatic cholesterol synthesis (better suited to statin therapy) or intestinal absorption (better suited to ezetimibe or other absorption inhibitors).
These tools refine strategy — they do not define risk. ApoB defines the risk. Subfractions and sterols help us understand the mechanism and tailor the response.
Metabolic Health Accelerates ASCVD
Two patients with identical apoB can have very different cardiovascular trajectories depending on their metabolic health. This is one of the most important and under-appreciated points in clinical lipid management.
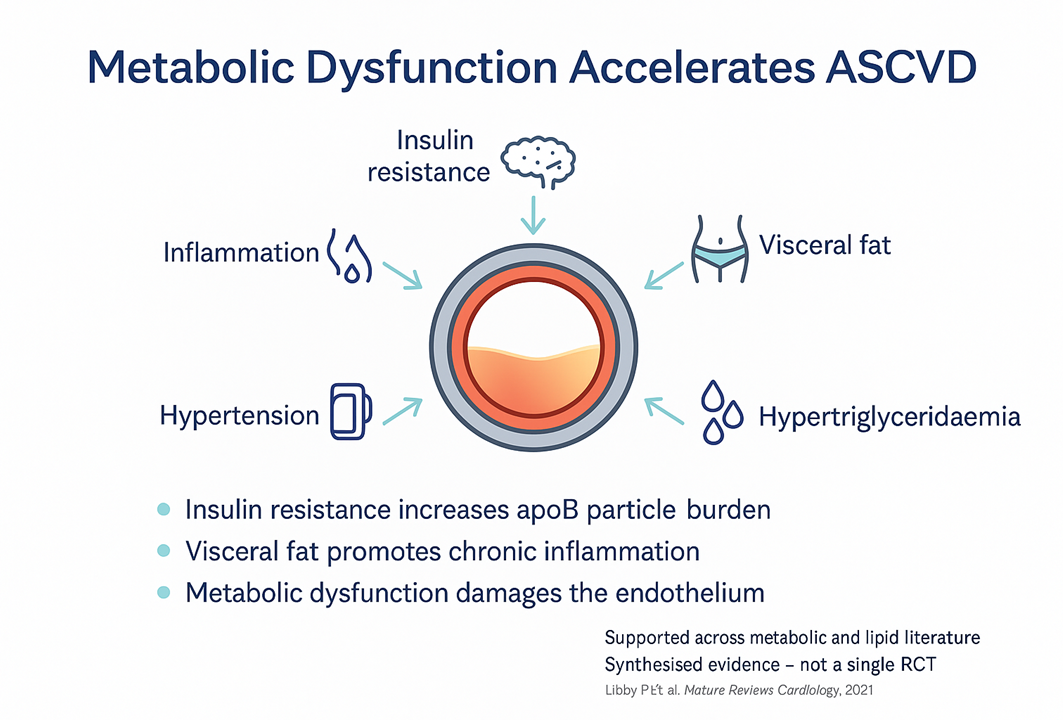
Insulin resistance increases VLDL production and flux through the liver, which raises apoB particle burden and shifts the LDL phenotype toward small, dense particles that are more readily retained in the arterial wall. Visceral adiposity drives chronic low-grade inflammation. Hypertension damages the endothelium directly. Hypertriglyceridaemia signals an underlying metabolic disturbance that is rarely benign.
ApoB tells us about particle burden. Metabolic health determines how damaging that burden becomes. The two need to be assessed together, not in isolation. A patient with apoB at the 75th percentile but excellent metabolic health is in a different position to a patient with the same apoB and severe insulin resistance — even though the particle count is the same.
ApoB reflects particle burden. Metabolic health determines how damaging that burden becomes.
Both need to be assessed together.
Biomarkers and Imaging Answer Different Questions
Blood-based markers and coronary imaging answer different questions. Both are useful. They are complementary, not interchangeable.

Blood markers — apoB, Lp(a), and the broader lipid and metabolic profile — estimate future risk. They tell us about the trajectory: how many particles are circulating, how genetically loaded the patient is, and how quickly disease is likely to accumulate.
Imaging tells us what disease is already present. Coronary artery calcium (CAC) scoring detects calcified plaque burden — a marker of historic atherosclerotic activity. CT coronary angiography (CTCA) goes further: it visualises the artery wall directly, identifying both calcified and non-calcified (“soft”) plaque. CTCA is particularly useful in younger patients where significant disease may exist before calcification has developed, and in characterising the type of plaque present.
Used together, biomarkers and imaging give a complete picture: what risk is accumulating going forward, and what disease has already accumulated. Either one alone leaves a gap.
Plaque Biology Matters — Beyond Stenosis
One of the most important shifts in modern cardiology is moving away from a stenosis-centric view of coronary disease. The lesions that cause myocardial infarctions are often not the most stenotic ones. Plaque composition matters as much as plaque burden.
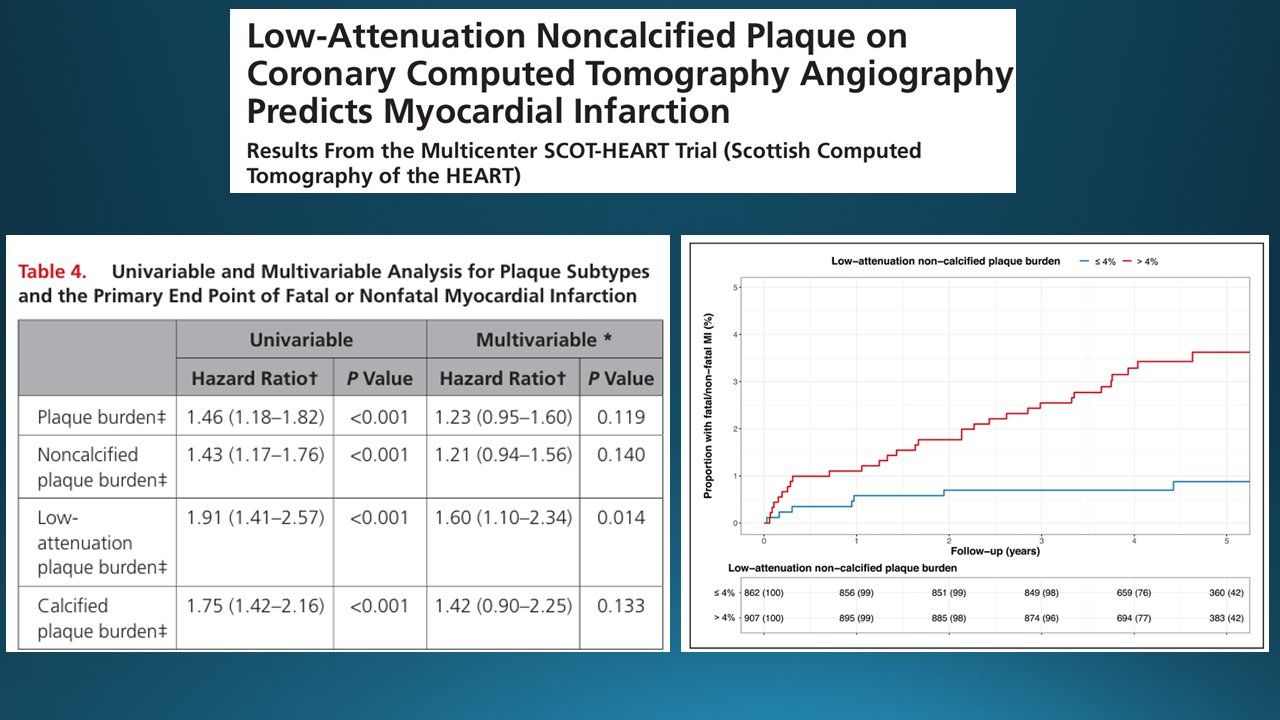
The SCOT-HEART trial provides the clearest contemporary evidence on this point. Williams and colleagues demonstrated that low-attenuation non-calcified plaque on CT coronary angiography — a marker of lipid-rich, vulnerable plaque — predicts myocardial infarction independent of stenosis severity, calcium score, and traditional risk factors. Patients with greater than 4% low-attenuation plaque burden had a substantially higher rate of fatal or non-fatal MI than those below that threshold [8].
This data explains several clinical patterns we have all seen: the patient with a normal stress test who has a myocardial infarction weeks later, the minimal luminal narrowing that produces a major event, and the limitation of stenosis-based thinking when applied to plaque biology.
Plaque biology, not just plaque obstruction, drives events. This is why CTCA (which visualises plaque composition rather than just luminal narrowing) has become an increasingly important tool in risk assessment.
Early vs Late Intervention: The ApoB × Time Framework
If ASCVD is fundamentally an exposure disease, then the timing of intervention has biological consequences that go beyond the magnitude of intervention. Late intervention flattens the exposure curve. Early intervention shifts the entire curve downward.
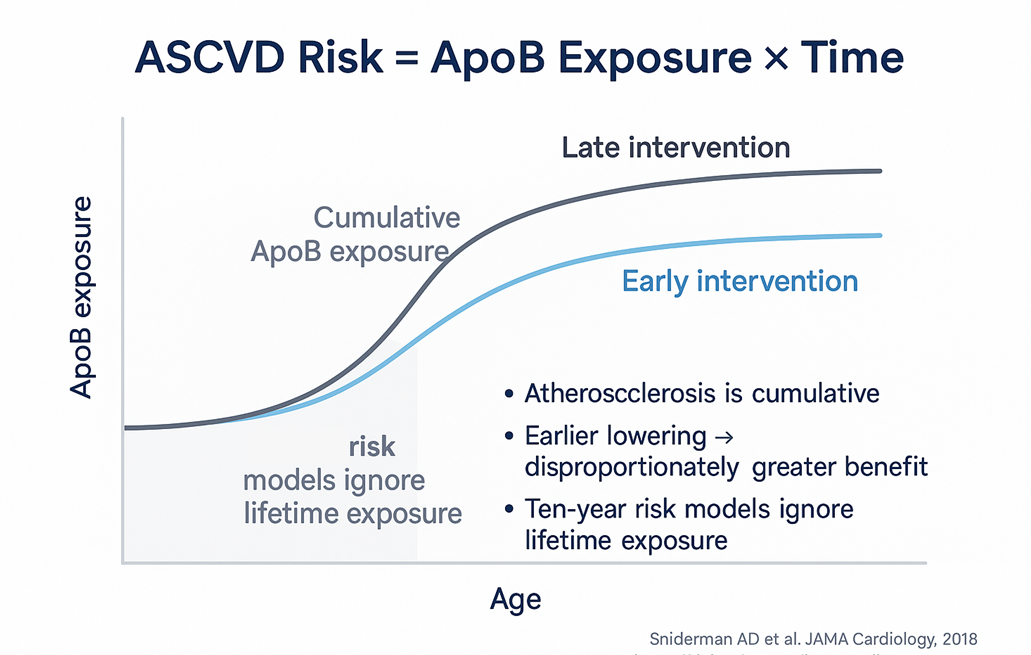
The Mendelian randomisation literature is consistent on this point. Genetic variants that produce lifelong modest reductions in LDL-C confer risk reductions roughly three-fold greater per mmol/L than the same magnitude of LDL-C reduction achieved pharmacologically in midlife [1, 2]. The biology is non-linear in time. Earlier intervention produces disproportionately greater benefit because the disease never accumulates in the first place — you are not trying to slow a process that is already advanced.
This does not mean treating every 30-year-old with a statin. It means understanding that the choice is rarely “intervene now” versus “wait” — it is more often “intervene now in a way proportionate to risk” versus “intervene later in a way that cannot recover the cumulative exposure already incurred.” Lifestyle interventions — nutrition, training, sleep, stress management — are the foundation of early intervention, with pharmacology added when the biological risk warrants it.
Late intervention flattens the curve but cannot erase prior exposure.
Early intervention shifts the entire exposure curve downward.
The pharmacological evidence base supporting LDL-C and apoB lowering is now substantial: statins (CTT meta-analysis [9]), ezetimibe (IMPROVE-IT [10]), PCSK9 monoclonal antibodies (FOURIER [11]), bempedoic acid in statin-intolerant patients (CLEAR Outcomes [12]), and inclisiran (ORION-10 and ORION-11 [13]). The selection between these agents depends on the individual patient — their tolerability, baseline particle burden, and the magnitude of reduction required — but the underlying principle is consistent: lowering apoB lowers events.
How This Translates Clinically
All of the above is theoretical until it shapes clinical practice. At Progressive Longevity Clinic, the framework above translates into a structured approach.
1. Measure Causal Risk
ApoB and Lp(a), alongside the standard lipid panel. ApoB tells us how many atherogenic particles are circulating now. Lp(a) tells us about lifetime genetic loading. Both inform the risk trajectory in ways that LDL-C alone cannot.
2. Assess Biological Accelerators
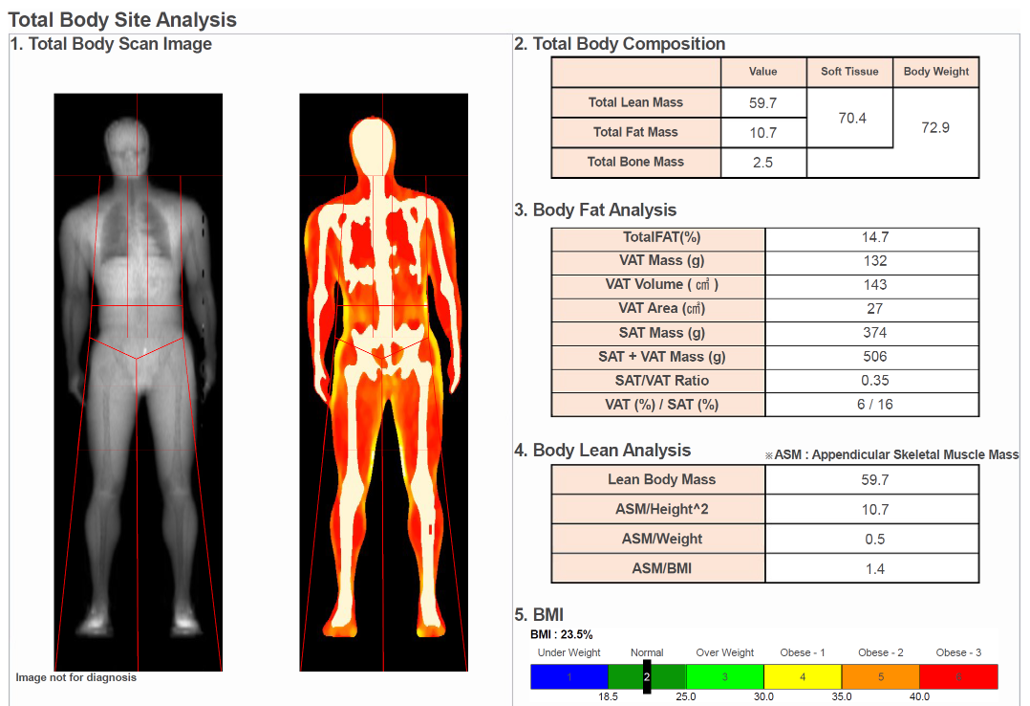
Insulin resistance markers (often including fasting insulin and HOMA-IR), visceral fat assessment (DEXA where appropriate), blood pressure profile, and inflammation (hsCRP). These determine how damaging any given particle burden is likely to be in this individual.
3. Identify Existing Disease When Appropriate
CAC scoring and CTCA when clinically indicated. Imaging is not for everyone, but for the right patient — typically those with elevated risk markers, a strong family history, or where the management decision hinges on whether disease is already present — it provides information that biomarkers alone cannot.
4. Intervene Early and Proportionately
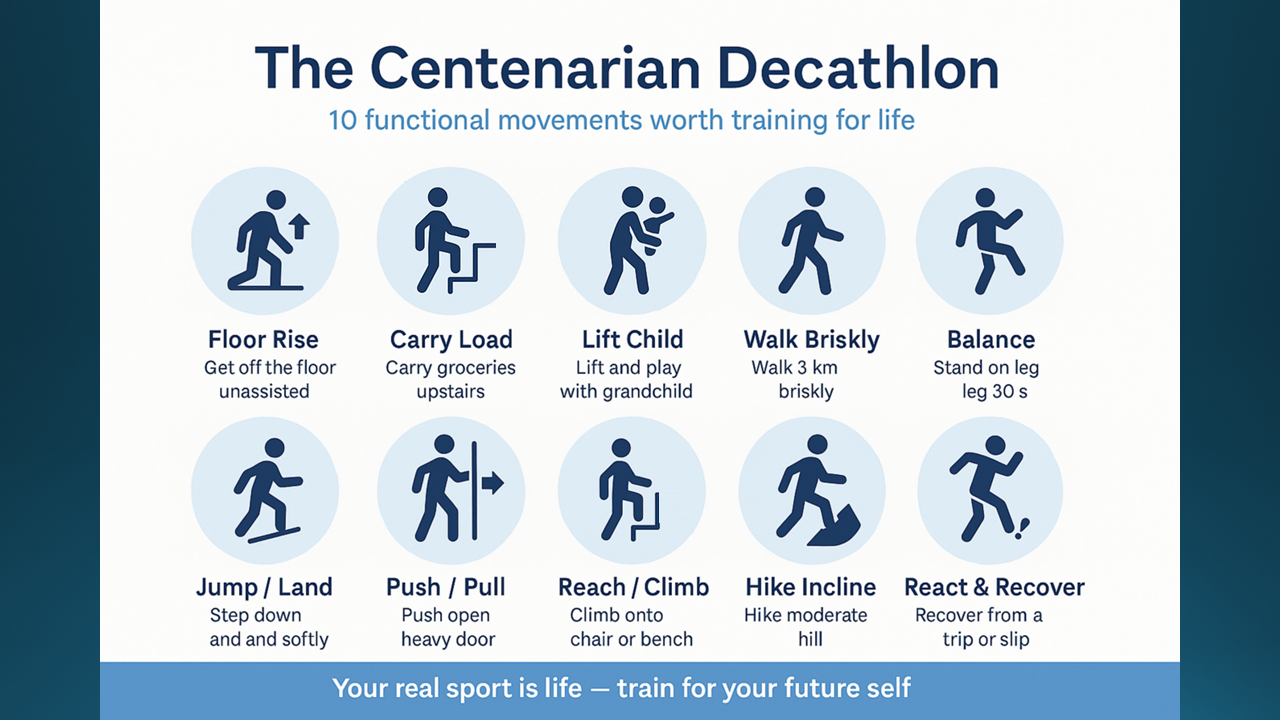
Exercise is foundational. Lifestyle is therapeutic. Medication is a tool, not a failure. The intervention package is built around the individual’s particular risk profile, biological accelerators, and personal preferences — documented and reviewed against defined endpoints.
The goal is not zero risk. Zero risk is not biologically achievable.
The goal is delaying disease long enough that it never becomes clinically relevant within the patient’s lifetime. That is a different objective — and a more honest one — than aspiring to a number on a calculator.
Summary: Reframing ASCVD as Cumulative Exposure

Across this article, the same threads keep recurring. ASCVD is a long disease, and the tools we routinely use are short-window tools. Cholesterol is the cargo; apoB-containing particles are the vehicle. Risk is cumulative, not binary. Disease begins decades before events occur. ApoB outperforms LDL-C as a risk marker, particularly when the two disagree. Lp(a) is a once-in-a-lifetime measurement that meaningfully shifts risk for one in five patients. Plaque biology, not just stenosis, drives events. And early, proportionate intervention is biologically rewarded out of proportion to the magnitude of the intervention itself.
None of this is to suggest that current cardiology is wrong. It is to suggest that current cardiology was built around managing established disease and reducing 10-year event rates — a useful but partial framework. Longevity-oriented cardiovascular medicine extends that framework upstream, asking how disease accumulates and where intervention can be made earlier with greater lifetime benefit.
The patient in front of us is rarely a 10-year risk score. They are a biological trajectory in motion- and one that we can meaningfully influence.
Interested in evidence-based cardiovascular risk assessment that goes beyond the standard lipid panel?
At Progressive Longevity Clinic, we work through ApoB, Lp(a), metabolic accelerators, and plaque imaging where appropriate — with proportionate, individualised intervention.
Visit our website to learn more about our Sport & Exercise Specialist-Led Longevity Medicine approach.
References
1. Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631-2639. DOI: 10.1016/j.jacc.2012.09.017
2. Ference BA, Bhatt DL, Catapano AL, et al. Association of genetic variants related to combined exposure to lower low-density lipoproteins and lower systolic blood pressure with lifetime risk of cardiovascular disease. JAMA. 2019;322(14):1381-1391. DOI: 10.1001/jama.2019.14120
3. Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B particles and cardiovascular disease: a narrative review. JAMA Cardiol. 2019;4(12):1287-1295. DOI: 10.1001/jamacardio.2019.3780
4. Marston NA, Giugliano RP, Melloni GEM, et al. Association of apolipoprotein B-containing lipoproteins and risk of myocardial infarction in individuals with and without atherosclerosis: distinguishing between particle concentration, type, and content. JAMA Cardiol. 2022;7(3):250-256. DOI: 10.1001/jamacardio.2021.5083
5. Libby P. The changing landscape of atherosclerosis. Nature. 2021;592(7855):524-533. DOI: 10.1038/s41586-021-03392-8
6. Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B particles and cardiovascular disease: a narrative review. JAMA Cardiol. 2019;4(12):1287-1295. DOI: 10.1001/jamacardio.2019.3780
7. Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;69(6):692-711. DOI: 10.1016/j.jacc.2016.11.042
8. Williams MC, Kwiecinski J, Doris M, et al. Low-Attenuation Noncalcified Plaque on Coronary Computed Tomography Angiography Predicts Myocardial Infarction: Results From the Multicenter SCOT-HEART Trial. Circulation. 2020;141(18):1452-1462. DOI: 10.1161/CIRCULATIONAHA.119.044720
9. Cholesterol Treatment Trialists' (CTT) Collaboration. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380(9841):581-590. DOI: 10.1016/S0140-6736(12)60367-5
10. Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes (IMPROVE-IT). N Engl J Med. 2015;372(25):2387-2397. DOI: 10.1056/NEJMoa1410489
11. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease (FOURIER). N Engl J Med. 2017;376(18):1713-1722. DOI: 10.1056/NEJMoa1615664
12. Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients (CLEAR Outcomes). N Engl J Med. 2023;388(15):1353-1364. DOI: 10.1056/NEJMoa2215024
13. Ray KK, Wright RS, Kallend D, et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol (ORION-10 and ORION-11). N Engl J Med. 2020;382(16):1507-1519. DOI: 10.1056/NEJMoa1912387
14. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551-561. DOI: 10.1161/01.ATV.15.5.551
15. Stary HC, Chandler AB, Glagov S, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89(5):2462-2478. DOI: 10.1161/01.CIR.89.5.2462
16. Newby DE, Adamson PD, Berry C, et al. Coronary CT Angiography and 5-Year Risk of Myocardial Infarction (SCOT-HEART). N Engl J Med. 2018;379(10):924-933. DOI: 10.1056/NEJMoa1805971
17. Mortensen MB, Falk E. Limitations of the SCORE-guided European guidelines on cardiovascular disease prevention. Eur Heart J. 2017;38(31):2259-2263. DOI: 10.1093/eurheartj/ehw568
18. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. DOI: 10.1093/eurheartj/ehz455
19. Greenland P, Blaha MJ, Budoff MJ, Erbel R, Watson KE. Coronary Calcium Score and Cardiovascular Risk. J Am Coll Cardiol. 2018;72(4):434-447. DOI: 10.1016/j.jacc.2018.05.027
20. Burgess S, Ference BA, Staley JR, et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018;3(7):619-627. DOI: 10.1001/jamacardio.2018.1470
This content is for educational purposes only and does not constitute individual medical advice. Cardiovascular risk assessment and intervention decisions should be individualised based on patient-specific factors, current clinical guidelines, and discussion with a qualified clinician. No patient-doctor relationship is established by viewing this material.
© 2026 Progressive Longevity Clinic, Sydney | Progressive Specialists